|
Clinical Genetics & Molecular Biology
GENETIC TESTING |
Genetic diseases:
A genetic disease or disorder is an abnormality in an individual's genetic material (genome). The abnormality could be a change in an individual’s DNA (mutation) or chromosomes (aberration)
Genetic diseases can be inherited or transmitted to next generation if the mutations are in the germ cells of the body (the cells involved in passing genetic information from parents to offspring). However, genetic diseases can also result from changes in DNA in somatic cells.
Genetic disorders can be categorized as (A) single-gene, (B) multifactorial, (C) chromosomal, and (D) mitochondrial.
(A) Single-gene (also called monogenic or Mendelian) disorder: This type is caused by changes or mutations that occur in the DNA sequence of one gene. A Gene is a functional unit of the genome consisting of a sequence of DNA that occupies a specific location on chromosome and code for proteins. When a gene is mutated so that its protein product can no longer perform its normal function, a disorder can result. Single-gene disorders are inherited in recognizable patterns: autosomal dominant, autosomal recessive, and X-linked.
(B) Multifactorial (also called polygenic or complex): This type is caused by a combination of environmental factors and mutations in multiple genes.
(C) Chromosomal: Chromosomes, discrete structures made up of DNA and protein, are carriers of genetic material. Abnormalities in chromosome structure as missing or extra copies or gross breaks and rejoining (translocations) can result in disease.
(D) Mitochondrial: This relatively rare type of genetic disorder is caused by mutations in the non-chromosomal DNA of mitochondria. Mitochondria are small round or rod-like organelles involved in cellular respiration and found in the cytoplasm of plant and animal cells. Each mitochondrion may have 5 to 10 circular pieces of DNA. |
Chromosomal analysis:
Chromosomal analysis / Cytogenetics involve the study of human chromosomes in health and disease. The chromosomes are visualized after staining, in light microscopy as they display a characteristic dark and light banding pattern according to the DNA content that is unique for individual chromosome. |
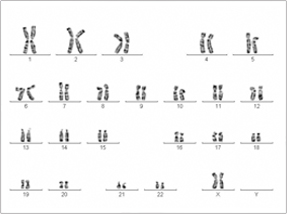 |
| Normal Female Karyotype |
|
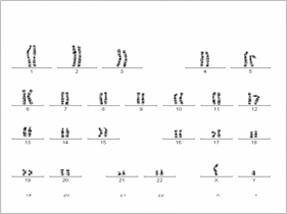 |
| Normal male Karyotype |
|
|
| It serves as an important laboratory diagnostic procedure in the field of prenatal diagnosis, in patients/families with history of mental retardation, multiple birth defects, abnormal sexual development, infertility or multiple miscarriages. The field also finds its application in malignancies and hematological disorders. |
Molecular diagnostics:
Molecular diagnostics assess your biological makeup at the genetic level. Sequencing the DNA can reveal variations between individuals which may represent a predisposition to develop a disease. Intricate pathogenesis pattern of a genetic disease can be addressed through molecular diagnostics. |
|
|
| Molecular diagnostics is the accurate method to screen the patients with ophthalmic genetic disorders. |
| It provides more information about the disease paving way for tailored treatments and personalized genomic medicine. |
| It can provide better disease management through genetic counseling. |
| Molecular diagnostics also helps patients to participate in treatment / clinical trials (research studies in people). |
Genetic counseling:
Genetic counseling is a service which elucidates the patients on genetic diseases. It is an understanding of facts behind the science of genetics. It is a cognitive operation of recuperating the quality of life. Our department dispenses this service for people with ocular diseases due to genetic defects.
List of genetic tests offered by our department: |
| Name of the test |
Condition for which the test is done |
| Chromosomal Study |
Chromosomal abnormalities |
| Rhodopsin (RHO) Gene Screening |
Retinitis pigmentosa & Congenital Stationary Night Blindness |
| RPE65 Gene Screening |
Retinitis Pigmentosa & Leber’s Congenital Amaurosis |
| CYP1B1 Gene Screening |
Peter’s anomaly, Congenital Glaucoma,
Primary and Juvenile Open Angle Glaucoma |
| MYOC Gene Screening |
Primary and Juvenile Open Angle Glaucoma |
| PAX6 Gene Screening |
Peter’s anomaly & Aniridia |
| Retinoschisis (RS1) Gene Screening |
Retinoschisis |
|
CHROMOSOMAL STUDY:
Chromosomal Study requires peripheral blood from which lymphocytes are extracted and cultured. Chromosomes in the cultured lymphocytes are studied either in interphase/metaphase/prometaphase stage. The metaphase chromosomes are preferred for analyzing the numerical/structural chromosomal abnormalities. |
 |
| Cytogenetics facility |
|
 |
| Cytogenetics facility |
|
|
We offer only postnatal analysis which include conditions like Down syndrome, Turner syndrome, Klinefelter syndrome, Wagner's syndrome, Microphthalmos, Anaphthalmos, Retinoblastoma, Aniridia, Axenfeld-rieger syndrome, Peters anomaly, Coloboma, Ptosis, Cri du chat syndrome etc.
1. Kumaramanickavel G, Mahboobu Subhani MSL, Rathnasabapathy V, Ramachandran MS (1987). A pilot study on the use of chromosomal analysis in the diagnosis of malignant effusions. South Indian Surgical Clinics; 1: 253-256.
2. Joseph B, Paul PG, Elamparithi A, Roy J, Vidhya A, Shanmugam MP, Kumaramanickavel G. Karyotyping in retinoblastoma—a statistical approach. Asian Pac J Cancer Prev. 2005 Oct-Dec; 6(4):468-71. PMID: 16435993[PubMed – indexed for MEDLINE]
RHODOPSIN GENE SCREENING:
Retinitis pigmentosa (RP) refers to a heterogeneous group of inherited diseases that result in progressive retinal degeneration, characterized by visual field constriction and night blindness. Mutations in the rhodopsin gene are a major contributor to various retinopathies such as retinitis pigmentosa and X-linked congenital stationary night blindness. Rhodopsin, also known as visual purple, is a biological pigment of the retina that is responsible for both the formation of the photoreceptor cells and the first events in the perception of light. A total of 103 mutations in rhodopsin are linked to RP to date and the phenotypes range from severe to asymptomatic. And there are reports stating, Vitamin A supplementation may confer therapeutic benefit by stabilizing certain mutant opsins through increased availability of the chromophore. Here, we screen the patients with retinitis pigmentosa for mutations in RHO gene by a standardized PCR based direct sequencing.
1. Kumaramanickavel G, Maw MA, Denton MJ, John S, Srikumari CRS, Orth U, Ralph O, Gal A. Missense rhodopsin mutation in a family with recessive RP. Nature Genetics; 1994:8: 10-11.
2. Gandra M, Anandula V, Authiappam V, Sundaramurthy S, Raman R, Bhattacharya S, Kumaramanickavel G. Retinitis pigmentosa: mutation analysis of RHO, PRPF31, RP1, and IMPDH1 genes in patients from India. Mol Vis. 2008 Jun 14; 14:1105-13. PMID: 18552984 [PubMed – indexed for MEDLINE]
|
|
RPE65 GENE SCREENING:
Leber Congenital Amaurosis (LCA) and Retinitis Pigmentosa (RP) are inherited degenerative diseases of the retina. LCA is characterized by bilateral congenital blindness. RP is characterized by night blindness, with age of onset varying from childhood to middle age, and progressing to constriction of the peripheral visual field and, eventually, to loss of central vision. Mutations in the RPE65 gene have been associated with Leber's congenital amaurosis type 2 (LCA2) and retinitis pigmentosa. The retinal pigment epithelium-specific 65 kDa (RPE65) protein is located in the retinal pigment epithelium and is involved in the conversion of all-trans retinol to 11-cis retinal during photo-transduction, which is then used in visual pigment regeneration in photoreceptor cells. Studies in animals and detailed human studies of photoreceptor layer integrity have led to gene therapy trials in LCA caused by mutations in RPE65. Jacobson et al in 2012 reported the safety and efficacy of those trials in 15 Children and adults followed up to 3 years where visual function improved in all patients to different degrees and improvements were localized to treated areas. Hence we provide a diagnostic testing for RPE65 mutations in patients with retinitis pigmentosa and Lebers congenital amaurosis. The testing involves a PCR based direct sequencing of all 14 exons in RPE65 gene in patients.
1. Kumaramanickavel G, Maw MA, Denton MJ, John S, Srikumari CRS, Orth U, Ralph O, Gal A. Missense rhodopsin mutation in a family with recessive RP. Nature Genetics; 1994:8: 10-11.
2. Kar B, John S, Kumaramanickavel G. Retinitis pigmentosa in India: a genetic and segregation analysis. Clinical Genetics;
1995: 47: 75-79.
3. Gu SM, Thompson DA, Srikumari CR, Lorenz B, Finckh U, Nicoletti A, Murthy KR, Rathmann M, Kumaramanickavel G, Denton MJ, Gal A. Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nature Genetics; 1997: 17:194 -197.
4. Finckh U, Xu S, Kumaramanickavel G, Schurmann M, Mukkadan JK, Fernandez T, John S, Weber JL, Denton MJ, Gal A. Homozygosity mapping of autosomal recessive retinitis pigmentosa locus (RP22) on chromosome 16p12.1-p12.3. Genomics; 1998 48:341-345.
5. Madhavan C, Bhende P, Lekha G. Vasanthi SB, Kumaramanickavel G. Retinitis Pigmentosa patients with Sickle Cell Disease and Dextrocardia and Situs Inversus Syndrome. Indian Journal of Ophthalmology; 2001:49:193-95.
6. Kumaramanickavel G, Selvaraj R, Rathanasabapathy V (1991). Retinitis pigmentosa: A genetic analysis. Afro-Asian J of Ophthalmology. Vol X: 20-25.
7. Joseph B, Anuradha S, Soumittra N, Vidhya A, Shetty NS, Uthra S, Kumaramanickavel G. RPE 65 gene: Multiplex PCR and Mutation screening in patients from India having retinal degenerative diseases. Journal of Genetics; 2002:81:1.
|
 |
| Pre-PCR facility |
|
CYP1B1 GENE SCREENING FOR GLAUCOMA:
Primary congenital glaucoma (PCG) is a recessively inherited disorder of the eye with variable penetrance. The trabecular meshwork and anterior chamber angle of the eye are affected, leading to the obstruction of aqueous outflow, increased intraocular pressure (IOP) and optic nerve damage. The onset of this disease is seen in the neonatal or early infancy period and, if untreated, results in an irreversible blindness. Genetic heterogeneity is well documented in PCG. At the GLC3A locus that harbors the human cytochrome P450 gene CYP1B1, more than 40 different mutations that cause PCG have been identified, indicating a high degree of allelic heterogeneity. CYP1B1 is proposed to be the major gene for disease causing mutations in primary congenital glaucoma. Cytochrome P450 1B1 is an enzyme that metabolizes a signaling molecule involved in eye development, possibly a steroid. Screening of the patients for CYP1B1 mutations will be helpful in assessing the carrier status of the gene and prenatal diagnosis.
1. Ramprasad VL, George R, Sripriya S, Nirmaladevi J, Vijaya L, Kumaramanickavel G. Molecular genetic analysis of a consanguineous South Indian family with congenital glaucoma: Relevance of genetic testing and counseling. Ophthalmic Genetics 2007 Jan-Mar; 28(1):17-24. |
|
MYOC GENE SCREENING FOR OPEN ANGLE GLAUCOMA:
Primary open-angle glaucoma is one of the leading causes of blindness in the world. It is a neurodegenerative disorder characterized by progressive excavation of the optic discs due to loss of retinal ganglion cells. It is usually associated with elevation of intraocular pressure (IOP). Based upon the age of diagnosis, primary open-angle glaucoma can be sub-classified to either juvenile-onset primary open-angle glaucoma (JOAG) or adult-onset primary open-angle glaucoma. JOAG is a relatively rare form of primary open angle glaucoma that occurs in children and young adults. Strong evidence indicates that genetic factors play a role in the pathogenesis of glaucoma. To date, three genes, namely myocilin (MYOC), optineurin (OPTN), and WD repeat-containing protein 36 (WDR36), have been reportedly linked to POAG. MYOC was the first gene to be identified as responsible for POAG. Almost 80 mutations have been found in MYOC. MYOC encodes the protein myocilin, which is normally secreted into the aqueous humor of the eye to control the intraocular pressure through its cytoskeletal action in the muscle tissue of the ciliary body. MYOC mutations, which cause myocilin to accumulate in the cells of the trabecular meshwork increasing the intra ocular pressure resulting in glaucoma. This genetic testing provides pre-symptomatic molecular diagnosis for members of a family and is useful for further genetic counseling with the family.
1. Sripriya S, Uthra S, Sangeetha R, George RJ, Hemamalini A, Paul PG, Amali J, Vijaya L, Kumaramanickavel G. Low frequency of Myocilin mutations in Indian POAG patients. Clinical Genetics Apr 2004 (4) 333 – 337. |
 |
| PCR Facility |
|
PAX6 GENE SCREENING FOR ANIRIDIA:
Aniridia is the absence of iris in the eye. Aniridia typically causes severe visual impairment; the major causative factor of this condition is foveal hypoplasia. Congenital aniridia is a rare ocular malformation that affects the development of multiple ocular structures. Aniridia is caused by a mutation in the paired box gene 6 (PAX6). A paired homeodomain transcription factor, PAX6, is a well-known regulator of eye development. Some sporadic cases have a risk of developing Wilms tumor as a part of WAGR (Wilms tumor, aniridia, genitourinary abnormalities, and mental retardation) which is caused by deletion of both PAX6 and Wilms’ tumor gene (WT1) in the 11p13 region. PAX6 protein acts as a "master control" gene for the development of eyes and other sensory organs, certain neural and epidermal tissues as well as other homologous structures usually derived from ectodermal tissues. This transcription factor is most noted for its use in the interspecifically induced expression of ectopic eyes and is of medical importance because heterozygous mutants produce a wide spectrum of ocular defects such as Aniridia in human. Numerous PAX6 mutations have been detected in aniridia patients and premature termination of the PAX6 protein is the most frequent type of mutation. The diagnostic testing for PAX6 mutation confirms the clinical diagnosis of aniridia. |
 |
| DNA sequencing facility |
|
RETINOSCHISIS GENE SCREENING:
X-linked juvenile retinoschisis is an inherited eye disorder that makes the inner layer of the retina split in a spoke-wheeled pattern. It usually occurs in both eyes at once. This disease affects more males than females. Fundus examination shows areas of schisis in the macula, sometimes giving the impression of a spoke wheel pattern. Affected males typically have vision of 20/60 to 20/120. The diagnosis of X-linked juvenile retinoschisis is based on fundus findings, results of electrophysiologic testing, and molecular genetic testing. RS1 is the only gene known to be associated with X-linked juvenile retinoschisis. Retinoschisin is a cell-cell adhesion protein which helps in maintaining the integrity of the retina. Mutation in the Retinoschisin (Rs1) gene results in defective protein leading to splitting of retinal layers (Schisis).Since the mode of inheritance is X-linked recessive, the genetic testing in the patient would help in assessing the carrier status, prenatal diagnosis and genetic counseling.
GENETIC COUNSELING:
Apart from chromosomal testing and molecular diagnostics our department also renders genetic counseling services through ophthalmic geneticist of the hospital. During the Genetic Counseling session a pedigree chart (family chart) of the patient is generated & the ophthalmologist accomplishes the following:
• Define the diagnosis.
• Comprehend the risk of transmission.
• Explicate the nature of the disease.
• Resolve the management of the disease.
1. Ramprasad VL, George R, Sripriya S, Nirmaladevi J, Vijaya L, Kumaramanickavel G. Molecular genetic analysis of a consanguineous South Indian family with congenital glaucoma: Relevance of genetic testing and counseling. Ophthalmic Genetics 2007 Jan-Mar; 28(1):17-24 |
| Required condition for specimen transport: |
|
| Name of the test |
Specimen |
| Chromosomal Study |
10 ml of heparinized blood to be transported at room temperature |
| Rhodopsin (RHO) Gene Screening |
| RPE65 Gene Screening |
| CYP1B1 Gene Screening |
| MYOC Gene Screening |
| PAX6 Gene Screening |
| Retinoschisis (RS1) Gene Screening |
|
|
|
|
|
|






